Immunology generates more molecular data than ever. Making sense of it as a whole is now the hard part:
- Gene expression profiles come from incompatible technologies.
- Animal models capture mechanisms that only partially translate to humans.
- Clinical outcomes emerge from biology that is distributed across tissues, cell types, and scales.
AI promised to help, but most biological models have learned only fragments of the picture: a single modality, a single species, a single resolution.
What immunology has been missing is not another specialized model, but a unifying foundation.
From fragmented observations to patient-level understanding
Immune-mediated diseases are complex, but they are not arbitrary. Across conditions like Crohn’s disease, psoriasis, and atopic dermatitis, the same biological programs reappear: conserved cytokine signaling pathways, shared genetic risk factors, recurring immune cell states. While their expression varies across tissues and species, the underlying mechanisms are conserved. This makes immunology a natural candidate for transfer learning, if models can bridge modalities and species.
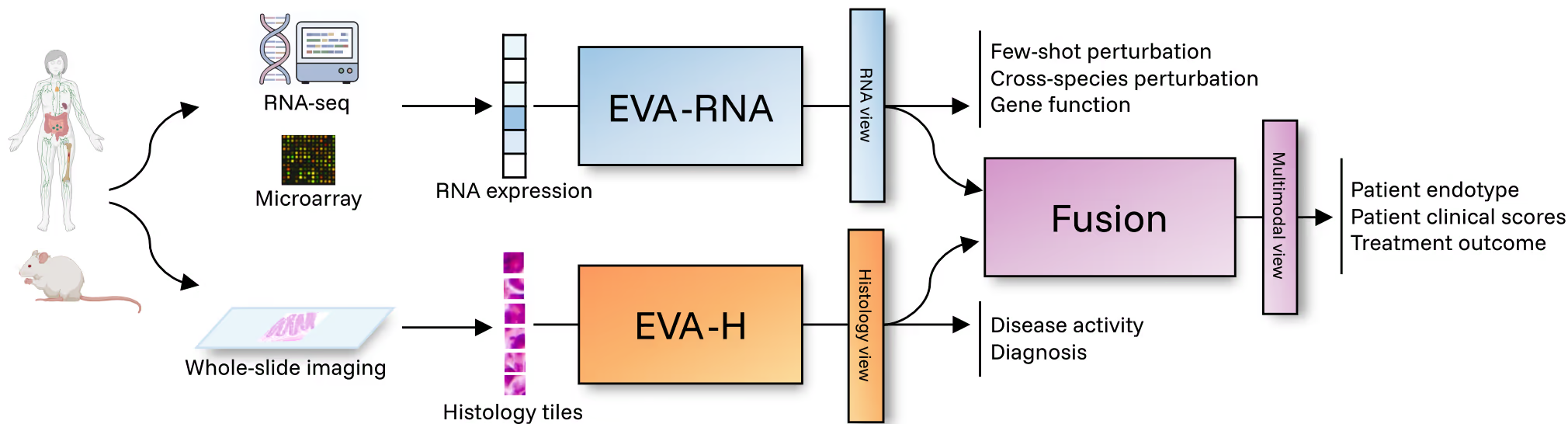
EVA was built around this premise. EVA is the first cross-species, multimodal foundation model designed specifically for immunology and inflammation. It integrates transcriptomics (microarray, bulk RNA-seq, single-cell–derived profiles) and histology (tissue imaging) from both human and mouse into unified, patient-level representations. This deliberate architectural choice forces the model to learn what is shared, not just what is convenient to measure. The result is a 440M-parameter model spanning 500K+ transcriptomics samples and 20M histology tiles across 50+ tissues and conditions. Its outcome is a single representation space where molecular profiles, tissue context, species differences, and disease states can be compared, queried, and reasoned about together.
Evaluation grounded in real R&D decisions
A recurring issue with biological AI is misalignment: current biological AI benchmarks reward models for solving irrelevant problems well, or the right problems on unreliable ground truth.
EVA was evaluated differently. We built a benchmark of nearly 40 tasks that mirror actual decisions across the drug discovery and development pipeline, from target discovery, to preclinical translation across species and clinical response prediction. Each task maps to a real decision a drug developer would face:
- Does perturbing this target move patients toward healthy state?
- Do molecular signals observed in mouse models translate to humans?
- Which patients are likely to respond to treatment?
Across all categories, EVA consistently outperforms existing foundation models. Critically, the model also avoids the failure mode where complex models are beaten by simple statistical baselines.
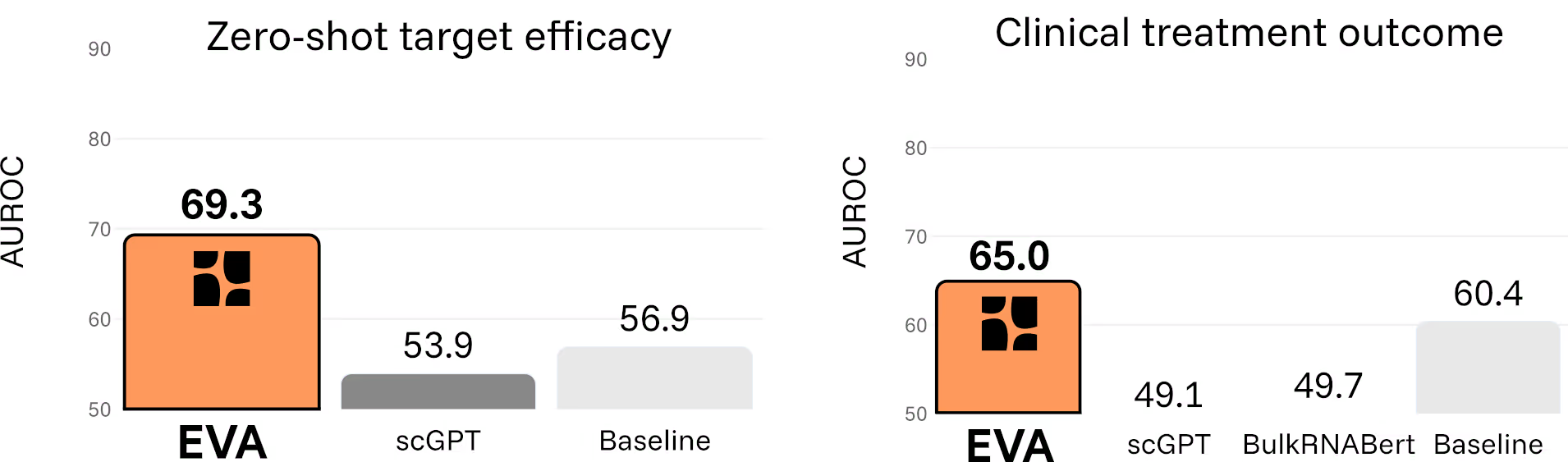
Doubling the success rate of clinical trials
One capability illustrates the shift particularly clearly. EVA can estimate whether a therapeutic target is likely to benefit patients with a given disease in zero-shot settings, without any task-specific training data. By simulating in silico gene perturbations directly on patient transcriptomes, the model predicts how molecular interventions reshape disease states.
Across six immuno-inflammatory diseases and 28 drugs, EVA distinguishes treatments with demonstrated clinical benefit from those without, capturing disease-specific effects rather than generic correlations. It correctly identifies, for example, why TNFα inhibition is effective in Crohn’s disease and psoriatic arthritis, but not in atopic dermatitis.
When framed as a binary decision, EVA’s predictions more than double the effective success rate compared to historical Phase II outcomes in this space.
Scaling laws and a roadmap forward
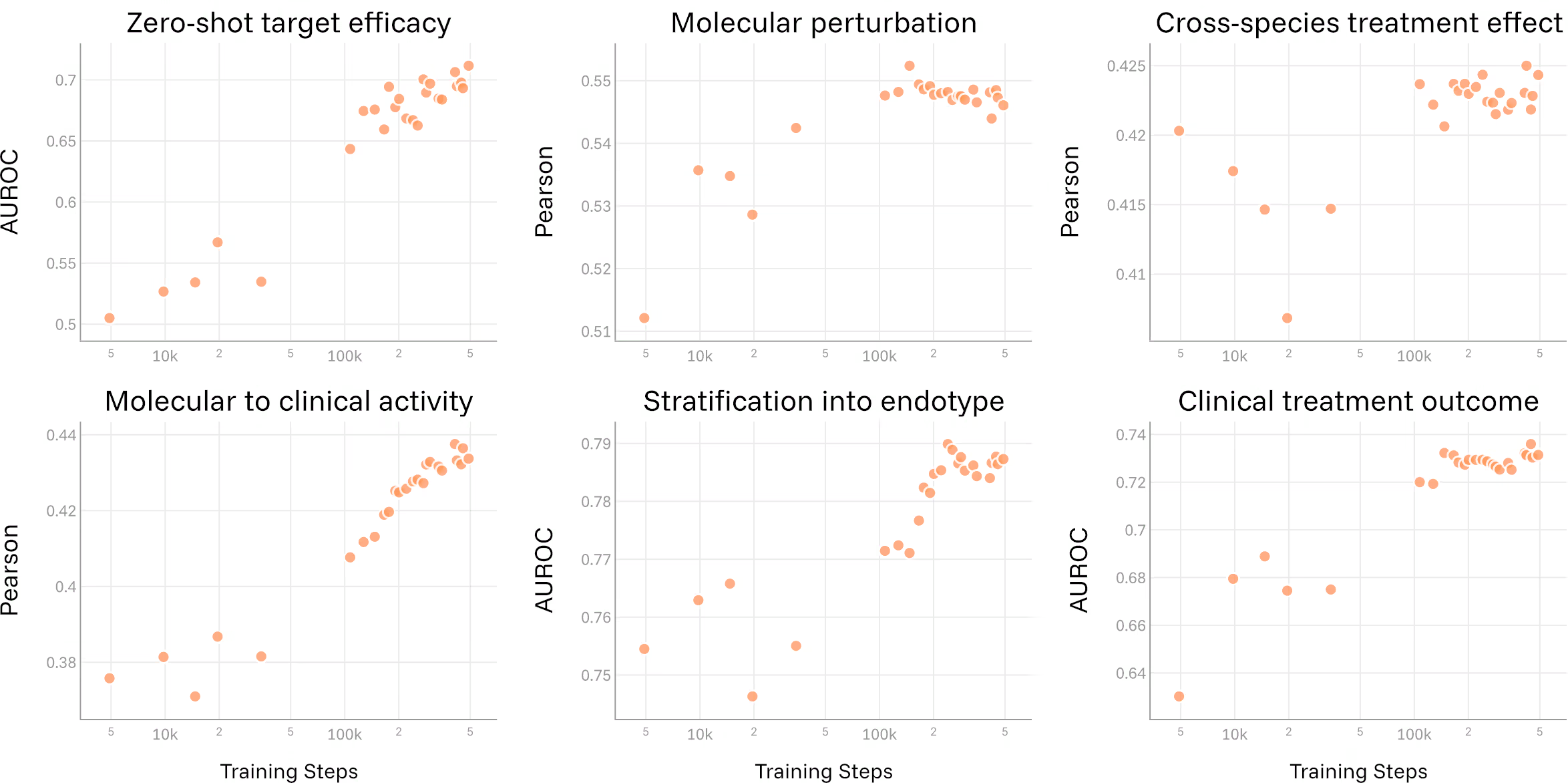
Another open question in the field has been whether biological foundation models benefit from scale in the same way language models do. EVA provides a clear signal: as model size and training compute increase, performance across translational tasks improves, with no sign of saturation yet. This establishes a principled path forward, scaling to more data, more tissues, and more conditions predictably improves the model's ability to answer translational questions, and we haven't hit the ceiling yet.
In immunology, scale is not excess. It is what allows the model to absorb heterogeneity without collapsing it.
Learning biology, not just patterns
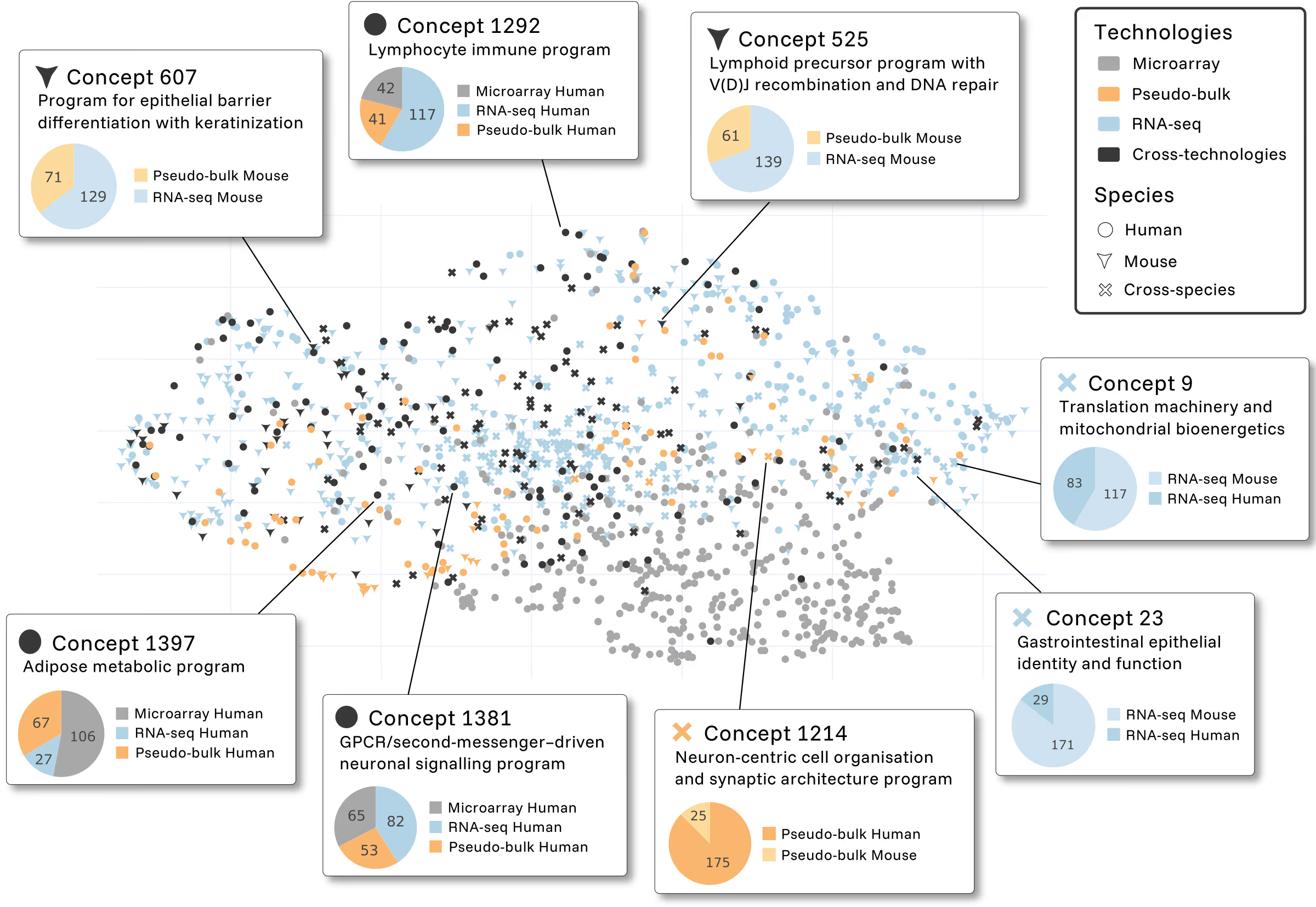
Foundation models are often criticized as black boxes. To address this directly, we applied mechanistic interpretability techniques to EVA’s representations.
What emerges are coherent biological programs: lymphocyte immune signatures, epithelial differentiation pathways, neuronal organization modules… concepts that activate consistently across species and technologies. These features are a map to known biology, providing trust in a model that shows what it has learned, without any supervision.
Opening the model to the community
EVA was built by the Scienta team in Paris with a single objective: to bring foundation models to the questions that matter most in immunology drug R&D.
As we continue to address this challenge, we are excited to introduce an open version of EVA for transcriptomics. The full technical details are available in our preprint, and the model can be accessed via Hugging Face.
This is not the end state.
It is the foundation.
{{CTA-1}}